
PGT Max 1
GIỚI THIỆU VỀ PGT MAX 1
| Danh mục kỹ thuật: - Xét nghiệm giải trình tự gen - Xét nghiệm đột biến gen gây dị tật bẩm sinh |
Xét nghiệm PGT MAX 1 (PGT phân giải cao) là xét nghiệm phân tích di truyền trước chuyển phôi với phiên bản nâng cấp về công nghệ phân tích kết quả giải trình tự gen so với xét nghiệm PGTest thông thường.
PGT MAX 1 giúp lựa chọn được phôi tối ưu hơn có chất lượng tốt về di truyền, làm tăng khả năng đậu thai khi thực hiện thụ tinh trong ống nghiệm (IVF), giúp cho trẻ sinh ra được khỏe mạnh, không mắc phải những bất thường di truyền đã sàng lọc.
| ƯU ĐIỂM VƯỢT TRỘI của PGT MAX 1: |
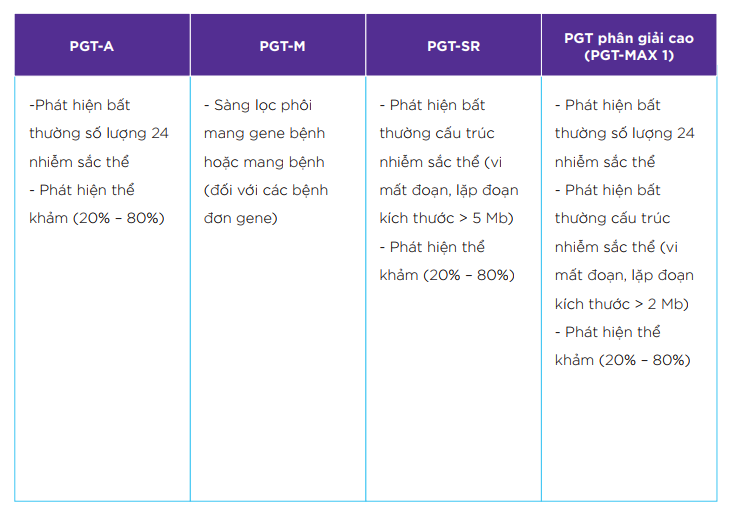
| Phát hiện bất thường số lượng 24 nhiễm sắc thể |
| Phát hiện thể khảm (20% – 80%) |
| Phát hiện bất thường cấu trúc, vi mất đoạn có kích thước > 5 Mb |
+ Mất đoạn 22q11.2 (liên quan đến hội chứng DiGeorge) + Mất đoạn 1p36 (liên quan đến hội chứng mất đoạn 1p36) |
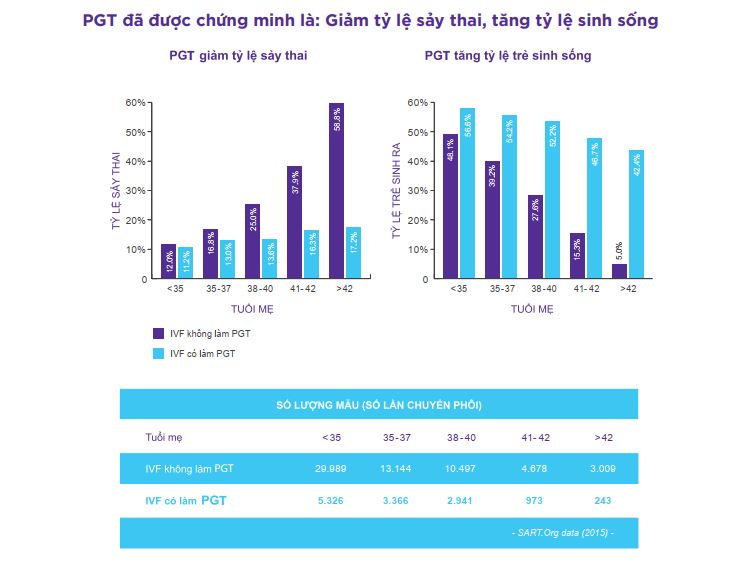
Ý NGHĨA XÉT NGHIỆM PGT MAX 1
Xét nghiệm PGT MAX 1 hỗ trợ chọn lọc các phôi không bị lệch bội 24 nhiễm sắc thể, không mang bất thường thêm/mất đoạn nhiễm sắc thể, hỗ trợ cho quá trình chọn lọc phôi được tối ưu hóa trước khi thực hiện chuyển phôi.
So với các xét nghiệm PGTest trước đây (PGT-A, PGT-SR) chỉ phát hiện được các vi mất đoạn có kích thước > 5Mb, xét nghiệm PGT MAX 1 phát hiện được thêm 2 hội chứng vi mất đoạn phổ biến có kích thước nhỏ tới 2Mb, liên quan tới những hội chứng di truyền với tỉ lệ trẻ sinh ra mắc bệnh ở mức khá cao là: Hội chứng DiGeorge & Hội chứng mất đoạn 1p36.
Hội chứng DiGeorge:
- Là hội chứng được mô tả lần đầu tiên vào năm 1968 bởi bác sĩ người Mỹ Angelo DiGeorge.
- Chiếm tỷ lệ khoảng 1/4000 trẻ được sinh ra, phổ biến thứ 2 sau hội chứng Down trong các hội chứng di truyền ở trẻ sơ sinh [1], [2].
- Hội chứng DiGeorge về kinh điển được mô tả do tổn thương vi mất đoạn nhánh dài nhiễm sắc thể 22 (microdeletion 22q11.2). Dạng đột biến này chiếm khoảng 90% các trường hợp được chẩn đoán hội chứng DiGeorge [3].
5 dấu hiệu thường gặp của Hội chứng DiGeorge:
- Dị tật tim bẩm sinh
- Hở hàm ếch
- Thiếu sản tuyến cận giáp dẫn đến nồng độ canxi trong máu thấp (gây động kinh)
- Nhiễm trùng
- Khuôn mặt đặc trưng là tai thấp, mắt rộng, hàm nhỏ, có rãnh hẹp ở môi trên
| Tài liệu tham khảo:
|
Nghiên cứu lâm sàng cho Hội chứng DiGeorge:
- Nghiên cứu đăng tải trên National Library of Medicine (NIH) với đề tài: “Một nghiên cứu dựa trên dân số về mất đoạn 22q11.2: Kiểu hình, Tỷ lệ mắc bệnh và Đóng góp vào các dị tật bẩm sinh lớn trong quần thể”
- Thực hiện trên 255 849 ca sinh:
- Tỉ lệ mắc phải của hội chứng này ở khoảng 1/6000 ở quần thể người da trắng, da đen và châu Á và khoảng 1/3800 ở quần thể người gốc Tây Ban Nha.
| Tài liệu tham khảo: Botto LD, May K, Fernhoff PM, Correa A, Coleman K, Rasmussen SA, Merritt RK, O'Leary LA, Wong LY, Elixson EM, Mahle WT, Campbell RM. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. |
- Một nghiên cứu đăng trên Tạp chí Phụ sản (VAGO) với đề tài: “Bất thường nhiễm sắc thể trên thai nhi dị tật tim bẩm sinh”
- Ở 92 thai phụ có thai nhi bất thường tim, được chọc hút dịch ối làm xét nghiệm nhiễm sắc thể đồ: kết quả cho thấy 2/92 trường hợp vi mất đoạn 22q11.2 (hội chứng DiGeorge)
- Bệnh tim bẩm sinh (BTBS) là những bất thường trong cấu trúc tim và các mạch máu trong khi mang thai ở tháng thứ 2-3 của thai kỳ, có tỷ lệ 14/1000 trẻ đẻ ra sống.
| Tài liệu tham khảo: https://vjog.vn/journal/article/view/699 Bùi Hải Nam(1), Trần Danh Cường(2) |

Hội chứng mất đoạn 1p36
- Mất đoạn 1p36 là một hội chứng điển hình gây ra các vấn đề về chậm phát triển trí tuệ. Hội chứng mất đoạn 1p36 có tỷ lệ mắc khoảng 1/5000 đến 1/10.000 ở trẻ sơ sinh
Biểu hiện của phần lớn những người mắc hội chứng Mất đoạn 1p36:
- Không nói được hoặc chỉ nói được một vài từ
- Có bất thường cấu trúc não, động kinh xảy ra ở hơn một nửa bệnh nhân mắc hội chứng này
- Có thể kèm theo nhược cơ và khó nhai nuốt (dysphagia)
Triệu chứng điển hình:
- Kích thước đầu mất cân đối, mắt sâu, lông mày thằng, phần giữa mặt lõm xuống (midface hypoplasia), mũi to;
- Khoảng cách mũi và miệng rộng (philtrum); cằm nhọn; tai có hình dạng bất thường.
- Có thể có vấn đề nhìn và nghe.
- Một số người có bất thường về hệ xương, tim, hệ tiêu hóa, thận hoặc cơ quan sinh dục.
Nghiên cứu lâm sàng cho Hội chứng mất đoạn 1p36:
- Nghiên cứu đánh giá: một tỷ lệ lớn trường hợp mất đoạn 1p36 không phải do di truyền. Nó là hậu quả do mất đoạn nhiễm sắc thể xảy ra ngẫu nhiên trong quá trình tạo giao tử (tinh trùng hoặc trứng) hoặc trong quá trình phát triển sớm của thai nhi.
- Người mắc hội chứng mất đoạn 1p36 mà do nhận được một chuyển đoạn không cân bằng sẽ bị thiếu vật chất di truyền ở cánh ngắn của nhiễm sắc thể 1 dẫn tới ảnh hưởng sau sinh và các vấn đề sức khỏe đặc trưng của hội chứng này.
| Tài liệu tham khảo: Abrantes, S. Sardinha, et al. "204 1p36 Deletion syndrome–A case report." European Journal of Obstetrics and Gynecology and Reproductive Biology 270 (2022): e14-e15. |

- Các đặc điểm khuôn mặt của một cô gái mắc hội chứng mất đoạn terminal 1p36
Những bức ảnh này thể hiện một số đặc điểm trên khuôn mặt là điển hình của trẻ em mắc hội chứng mất đoạn 1p36, bao gồm lông mày thẳng, cầu mũi rộng và cằm nhọn.
Ảnh được chụp vào lúc (a) 1 năm 8 tháng, (b) 2 năm 3 tháng, (c) 4 năm, (d) 7 năm, (e) 7 năm 11 tháng và (f) 10 năm 3 tháng tuổi.

| Tài liệu tham khảo: Jordan, V. K., Zaveri, H. P., & Scott, D. A. (2015). 1p36 deletion syndrome: an update. The application of clinical genetics, 8, 189–200. |
BẢNG TỔNG HỢP TỶ LỆ TRẺ MẮC BỆNH/TRẺ SINH RA VỚI CÁC HỘI CHỨNG DI TRUYỀN ĐẶC BIỆT

=> Từ các nghiên cứu lâm sàng về hai hội chứng di truyền trên, việc thực hiện sàng lọc bất thường di truyền trước chuyển phôi PGT MAX 1 là xét nghiệm rất cần thiết.
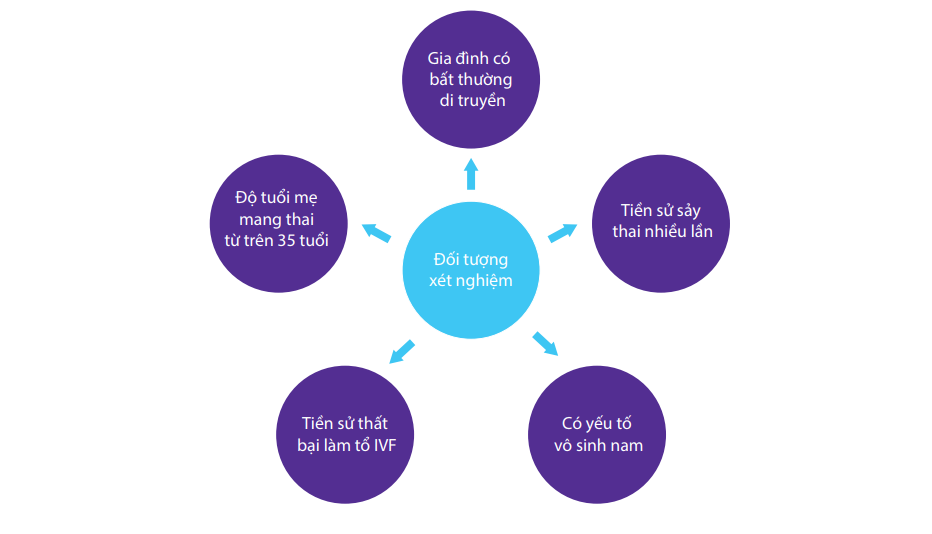
ĐỐI TƯỢNG NÊN THỰC HIỆN XÉT NGHIỆM PGT MAX 1
- Độ tuổi mẹ mang thai từ trên 35 tuổi
- Tiền sử sảy thai nhiều lần
- Tiền sử thất bại làm tổ IVF
- Có yếu tố vô sinh nam
- Gia đình có bất thường di truyền hoặc có con mang bất thường di truyền
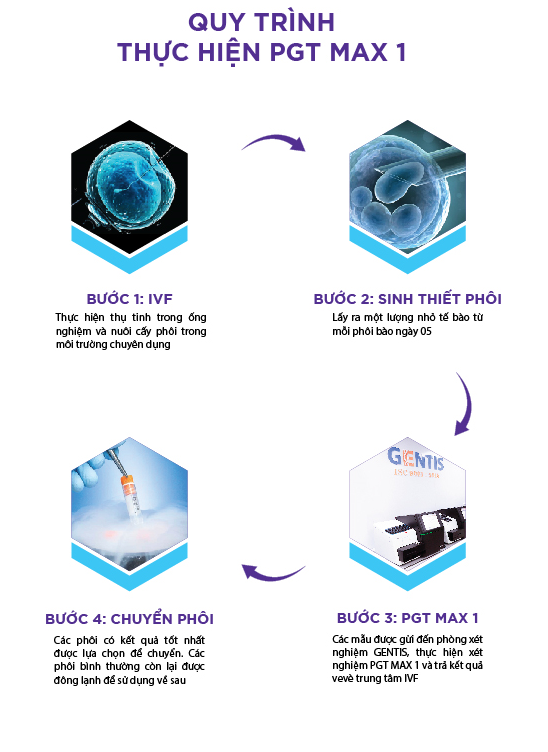
QUY TRÌNH THỰC HIỆN XÉT NGHIỆM PGT MAX 1
| Bước 1: Lấy mẫu sinh thiết phôi ngày 5 |
| Bước 2: Tách chiết ADN của mẫu tế bào phôi và khuếch đại bằng bộ Kit Veriseq PGS |
| Bước 3: Giải trình tự trên máy giải trình tự thế hệ mới NGS (Illumina) |
| Bước 4: Phân tích kết quả giải trình tự bằng phần mềm tin sinh chuyên dụng |
| Bước 5: Trả kết quả 5-7 ngày |
XÉT NGHIỆM PGT MAX 1 TẠI GENTIS
Trên thị trường hiện nay, GENTIS là đơn vị đầu tiên triển khai xét nghiệm PGT Max 1 với công nghệ update tiên tiến, cho phép phát hiện thêm các bất thường vi mất đoạn/ vi lặp đoạn (có kích thước >2mb) liên quan đến một số bệnh và hội chứng di truyền phổ biến.
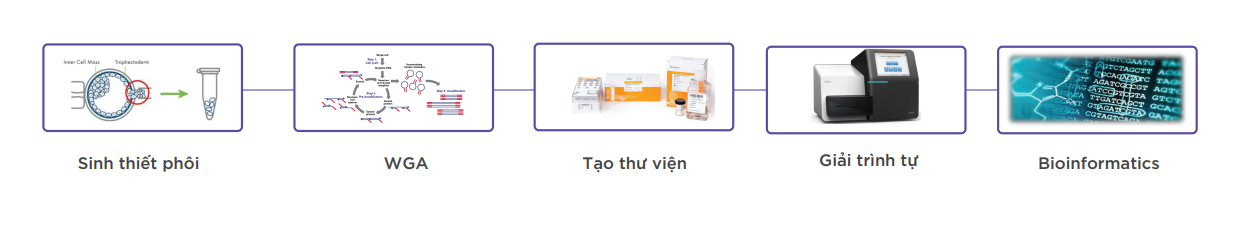
- Mẫu sử dụng: Mẫu sinh thiết phôi ngày 5
- Phương pháp sử dụng: Giải trình tự thế hệ mới NGS (Illumina) và phân tích kết quả giải trình tự bằng phần mềm tin sinh chuyên dụng để xác định lệch bội toàn bộ 24 NST và các bất thường thêm, mất đoạn NST.
- Thời gian trả kết quả: 5 – 7 ngày
- Công nghệ xét nghiệm:
- Sử dụng công nghệ giải trình tự của hãng Illumina, Hoa Kỳ và Phần mềm phân tích chuyên dụng
- Độ tin cậy cao với độ nhạy >99% và độ đặc hiệu >99%
- Độ phân giải lớn, phát hiện được những bất thường cấu trúc nhiễm sắc thể (mất đoạn, lặp đoạn > 5mb), đặc biệt phát hiện thêm các mất đoạn > 2Mb liên quan đến những hội chứng di truyền phổ biến như: hội chứng DiGeorge (mất đoạn 22q11.2) và hội chứng 1p36
ĐĂNG KÝ DỊCH VỤ TẠI GENTIS
Quý khách vui lòng điền thông tin bên dưới để được hỗ trợ,
tư vấn một cách tốt nhất!