-1366x0.jpg)
1, Bệnh Thalassemia là gì?
a. Thalassemia:
Thalassemia bắt nguồn từ sự kết hợp của hai từ Hy Lạp: Thalassa (nghĩa là biển “sea”, ở đây là Địa Trung Hải) và anemia (thiếu máu – “weak blood”). Một thuật ngữ nữa cũng được tìm thấy, tuy không thường xuyên là Cooley’s anemia (thiếu máu Cooley, tên của giáo sư Cooley Thomas, một bác sĩ nhi khoa Hoa Kỳ, người đầu tiên mô tả các đặc điểm lâm sàng của rối loạn này ở bệnh nhân gốc Ý vào năm 1925).
Thuật ngữ Thalassemia (còn gọi là tan máu bẩm sinh), đây là bệnh rối loạn máu được di truyền lặn trên nhiễm sắc thể thường, bệnh xảy ra do khiếm khuyết tổng hợp 1 hay nhiều chuỗi globin. Rối loạn này dẫn đến một số lượng lớn các tế bào hồng cầu bị phá hủy, dẫn đến thiếu máu. Bệnh Thalassemia được gọi tên theo chuỗi Globin bị khiếm khuyết. Gồm hai loại chính là Anpha Thalassemia và Beta Thalassemia.
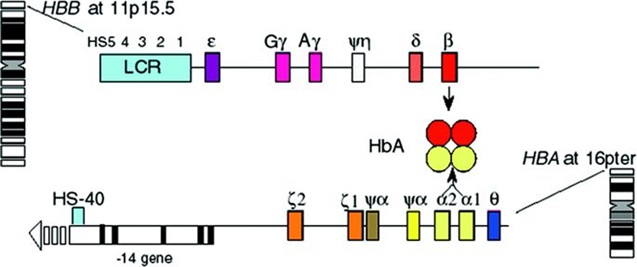
Hình 1: Vị trí và cấu trúc NST của các cụm gen của chuỗi alpha và beta Thalassemia. Trong đó, gen alpha Thalassemia (HBA gồm HBA1 và HBA2) nằm trên nhiễm sắc thể số 16; gen beta Thalassemia (HBB) nằm trên nhiễm sắc thể số 11.
b. Alpha Thalassemia:
Alpha Thalassemia gồm 4 gen (của hai bản sao HBA1 và HBA2) nằm trên NST 16. Đột biến alpha Thalassemia được gây ra bởi việc mất hoặc đột biến ở một hoặc nhiều trong bốn gen alpha globin. Đột biến gây ra sự giảm sản xuất alpha globin. Càng nhiều gen bị ảnh hưởng, cơ thể càng ít tạo ra alpha globin.

Hình 2: Các kiểu gen của alpha- Thalassemia dựa trên số gen anpha bị mất.
- Khi đột biến xảy ra ảnh hưởng ở cả hai loci, kiểu gen gọi là a-Thal1
- Khi đột biến xảy ra ảnh hưởng 1 locus, kiểu gen gọi là a-Thal2.
- Locus: vị trí của gen trên NST, loci: số nhiều của locus.
- Homozygous: đồng hợp tử; Heterozygous: Dị hợp tử
Bốn loại alpha Thalassemia khác nhau được phân loại theo số lượng gen bị ảnh hưởng và bao gồm:
• Silent Carrier State (thể ẩn- 1 gen bị ảnh hưởng): Những người chỉ có một gen alpha globin là người mang mầm bệnh ẩn (hay còn gọi là thể ẩn). Họ thường có nồng độ hemoglobin và các chỉ số hồng cầu bình thường nhưng có thể truyền gen bị ảnh hưởng cho con cái của họ. Những cá nhân này không có dấu hiệu hoặc triệu chứng và thường chỉ được xác định sau khi có con bị bệnh Thalassemia. Cách để xác định người mang gen thể ẩn là phân tích ADN Thalassemia.
• Alpha Thalassemia Trait (2 gen bị ảnh hưởng): các tế bào hồng cầu (RBCs) nhỏ hơn (microcytic) và nhạt hơn (hypochromic) so với bình thường, có MCV giảm và thiếu máu nhẹ. Dạng thiếu máu này không đáp ứng với chất bổ sung sắt. Chẩn đoán dạng alpha Thalassemia Trait thường được thực hiện bằng cách loại trừ các nguyên nhân khác gây thiếu máu vi mô. Kiểm tra xác nhận bằng phân tích ADN Thalassemia có thực hiện nhưng hiện nay vẫn chưa thường xuyên.
• Hemoglobin H (3 gen bị ảnh hưởng): Cơ thể giảm sản xuất chuỗi alpha globin gây ra sự dư thừa chuỗi beta, sau đó các chuỗi beta kết hợp thành nhóm 4 chuỗi beta, được gọi là HemoglobinH (HbH). Bệnh Hb H có thể gây thiếu máu từ trung bình đến nặng gây ra mệt mỏi, lách to, biến dạng xương. Các dấu hiệu và triệu chứng liên quan đến bệnh Hb H rất khác nhau. Một số cá nhân không có triệu chứng trong khi những người khác bị thiếu máu nghiêm trọng, cần được chăm sóc y tế thường xuyên. Bệnh HbH được tìm thấy thường xuyên nhất ở những người gốc Đông Nam Á hoặc Địa Trung Hải.
• Alpha Thalassemia Major (còn gọi là phù thai (hydrops fetalis), 4 gen bị ảnh hưởng). Đây là dạng alpha Thalassemia nghiêm trọng nhất. Ở dạng này, không có alpha globin được sản xuất, do đó, không có huyết sắc tố bình thường được sản xuất. Thai nhi ở dạng này bị thiếu máu sớm trong thai kỳ. Chúng giữ lại các chất lỏng dư thừa (hydropic - phù) và thường có tim và gan to. Chẩn đoán này thường được thực hiện trong những tháng cuối của thai kỳ khi siêu âm thai cho thấy thai nhi bị phù (hydrops fetalis). Cũng có những rủi ro cho người mẹ mang thai. Khoảng 80% thời gian, mẹ sẽ bị "nhiễm độc máu - toxemia " (protein trong nước tiểu, huyết áp cao, mắt cá chân và bàn chân sưng) và có thể bị chảy máu sau sinh nghiêm trọng (xuất huyết, băng huyết). Các thai nhi mắc bệnh alpha Thalassemia Major thường bị sảy thai, chết non hoặc chết ngay sau khi sinh. Trong những trường hợp rất hiếm, trẻ em mắc bệnh alpha Thalassemia đã sống sót qua truyền máu tử cung và chăm sóc y tế đặc biệt.
c. Beta Thalassemia:
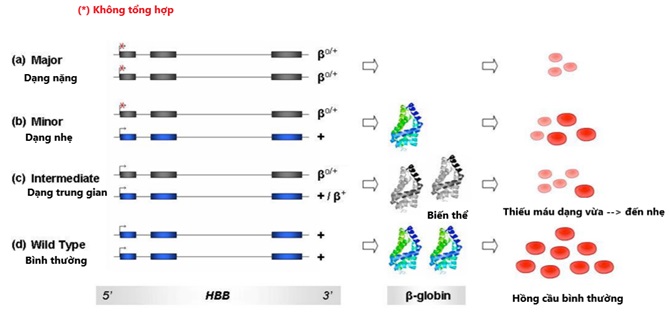
Hình 3: Sơ đồ thể hiện các kiểu hình của chuỗi. Các biến thể HBB được biểu thị bằng exon màu xám, kiểu bình thường được biểu thị bằng màu xanh. Các hồng cầu đỏ tươi đại diện cho kiểu hồng cầu bình thường; các hồng cầu màu hồng nhạt đặc trưng cho các kiểu thiếu máu.
Beta Thalassemia gồm 2 gen của bản sao HBB nằm trên NST 11. Đột biến beta Thalassemia được gây ra bởi đột biến ở một hoặc cả hai gen beta globin. Đã có hơn 250 đột biến được xác định, nhưng chỉ có khoảng 20 đột biến là phổ biến nhất. Mức độ nghiêm trọng của bệnh thiếu máu do beta Thalassemia phụ thuộc vào đột biến xuất hiện và sự giảm sản xuất beta globin (được gọi là beta+ Thalassemia) hay hoàn toàn không sản xuất (gọi là beta0 Thalassemia).
Các loại beta Thalassemia khác nhau bao gồm:
• Beta Thalassemia Trait hoặc Beta Thalassemia Minor. Các cá nhân mắc bệnh này có một gen bình thường và một gen bị đột biến, gây ra sự giảm nhẹ sản xuất beta globin. Họ thường không có vấn đề sức khỏe nào ngoài các tế bào hồng cầu nhỏ bất thường và thiếu máu nhẹ, có thể không đáp ứng với các chất bổ sung sắt. Con cái của họ có thể bị ảnh hưởng do di truyền.
• Bệnh Thalassemia Intermedia (dạng trung gian). Dạng này có hai gen bất thường, làm giảm sản xuất beta globin từ trung bình đến nghiêm trọng. Những người này có thể phát triển các triệu chứng muộn hơn so với những người mắc bệnh Thalassemia major (xem bên dưới) và thường có các triệu chứng nhẹ hơn. Những người dạng này có thể thỉnh thoảng cần truyền máu. Trong thể beta Thalassemia trung gian, bệnh nhân có Hb tiêu thụ năng lượng dưới 7 hoặc 8 gm/ dl do tan máu sâu (profound hemolysis) có thể tạo ra tầm vóc nhỏ, tăng cân kém, dễ bị nhiễm trùng, vàng da, vàng mắt và màng nhầy gây ra bởi sự gia tăng lượng bilirubin trong máu.
• Thalassemia Major (hay Cooley's Anemia). Đây là dạng beta Thalassemia nghiêm trọng nhất. Những người mắc dạng này có hai gen bất thường gây ra sự sụt giảm nghiêm trọng hoặc thiếu hoàn toàn việc sản xuất beta globin, ngăn chặn việc sản xuất một lượng đáng kể huyết sắc tố bình thường (Hb A). Tình trạng này thường xuất hiện trong vòng hai năm đầu đời và gây thiếu máu đe dọa tính mạng, tăng trưởng kém, các bất thường về xương trong giai đoạn ấu thơ và các triệu chứng xuất hiện trong hai năm đầu đời. Trẻ sơ sinh sẽ không phát triển nhanh, không tăng cân bình thường và dần dần trở nên xanh xao. Các vấn đề về ăn uống, tiêu chảy, khó chịu, sốt và bụng càng ngày càng to do lách to và nổi bật của xương gò má có xu hướng che khuất cơ sở của mũi và để lộ răng hàm trên, bọng mắt và dẫn đến xu hướng mắt xếch Mongoloid. Do đó, những người dạng này đòi hỏi phải truyền máu thường xuyên. Về lâu dài sẽ gây ra tình trạng ứ sắt. Nếu không được điều trị, lượng sắt dư thừa này có thể lắng đọng trong gan, tim và các cơ quan khác và có thể dẫn đến tử vong sớm do suy nội tạng. Do đó, các cá nhân trải qua truyền máu có thể cần điều trị thải sắt để giảm quá tải sắt.
Có thể tóm tắt phân loại trên bằng bảng dưới đây:
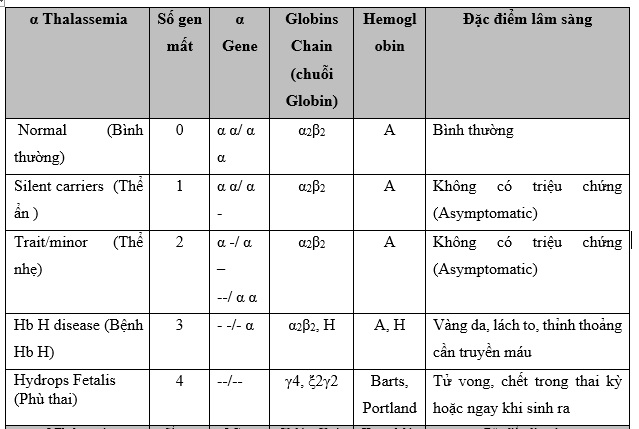
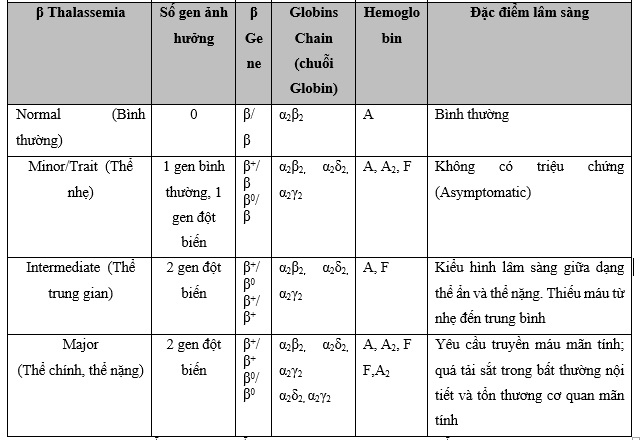
Bảng 1: Tổng hợp các kiểu gen Thalassemia và các đặc điểm lâm sàng tương ứng
2. Cơ chế di truyền của bệnh:
Thalassemia là một bệnh di truyền lặn, được di truyền từ bố mẹ cho các thế hệ sau. Theo quy luật di truyền, bố và mẹ mỗi người truyền một nửa vật chất di truyền cho con cái. Theo sơ đồ dưới đây, nếu cả một trong hai bố mẹ bình thường, người kia mang gen bệnh, tỷ lệ sinh con là 50% bình thường :50% con mang gen bệnh. Nhưng nếu cả hai bố và mẹ đều mang gen bệnh thì tỷ lệ sinh con là 25% bình thường: 50% mang gen bệnh: 25% bị bệnh. Do đó, nếu bố mẹ không có biểu hiện của bệnh thiếu máu hoặc biểu hiện dạng nhẹ nên đi xét nghiệm ADN Thalassemia. Dựa vào kết quả xét nghiệm của bố mẹ; bác sĩ chuyên môn sẽ tư vấn các hướng hỗ trợ sinh sản để sinh ra đứa bé khỏe mạnh, không mắc bệnh di truyền từ bố mẹ hoặc chỉ ở dạng nhẹ (không ảnh hưởng đến sức sống của trẻ). Một trong những hướng tiếp cận hiện đại (nếu cả hai bố mẹ có mang gen bệnh) là kỹ thuật PGD (preimplantation genetic screening- sàng lọc di truyền trước chuyển phôi).
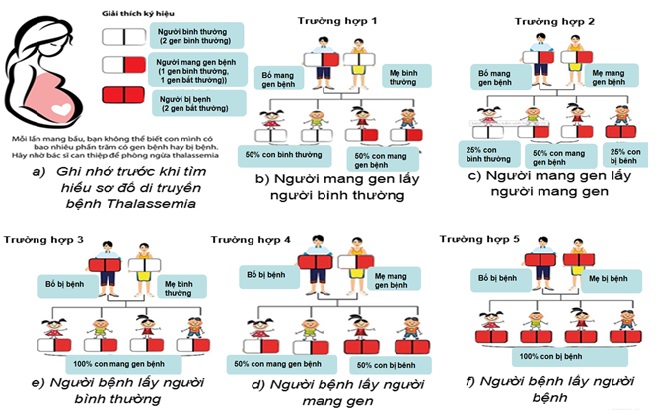
Hình 3: Sơ đồ di truyền từ bố mẹ cho con cái
3. Lịch sử phát hiện và vùng phân bố bệnh Thalassemia:
Thalassemia tràn lan khắp các khu vực Địa Trung Hải, Châu Phi, Trung Đông, tiểu lục địa Ấn Độ, Trung Á và Đông Nam Á. Điều này có khả năng liên quan đến tỷ lệ mắc bệnh sốt rét ở những khu vực này vì bệnh Thalassemia có thể làm tăng khả năng chống sốt rét. Ở những vùng này, tỷ lệ mắc bệnh Thalassemia có thể lên tới 10%. Bệnh có thể gặp ở cả nam và nữ với tỷ lệ mắc bệnh như nhau.
Bệnh Thalassemia là gánh nặng toàn cầu với ước tính rằng khoảng 280 triệu người mắc bệnh trên toàn thế giới, trong đó khoảng 439,000 người mắc với dạng nặng và nó dẫn đến 16,800 trường hợp tử vong trong năm 2015.Theo thống kê từ Liên Hiên Quốc, dân số Việt Nam tính đến ngày 21/07/2019 là 97.480.634 người (gần 98 triệu người).
Tại Việt Nam tỷ lệ người mắc Thalassemia khá cao (chiếm gần 11-13% dân số, ước tính khoảng 12 triệu người, tính cả người mắc bệnh và người mang gen bệnh). Theo số liệu thống kê tại GENTIS HCM, từ 1/11/2017 đến 30/07/2019 có 320 ca. Trong đó có 83 ca âm tính; 126 ca mắc alpha Thalassemia; 98 ca mắc beta Thalassemia; 13 ca vừa mắc alpha và beta (chủ yếu là các dị hợp alpha SEA kết hợp với beta E (G-A), CD17, CD41/42 hoặc dị hợp alpha 3.7 kết hợp với beta E, CD41/42).

Hình 4: Tổng hợp dữ liệu các ca Thalassemia thực hiện tại GENTIS HCM từ 1/11/2017 đến 30/07/2019. Trong đó alpha Thalassemia chiếm 39% (126/320); beta Thalassemia chiếm 31% (98/320); vừa mắc anpha, vừa mắc beta chiếm 4% (13/320); âm tính chiếm 26% (83/320).
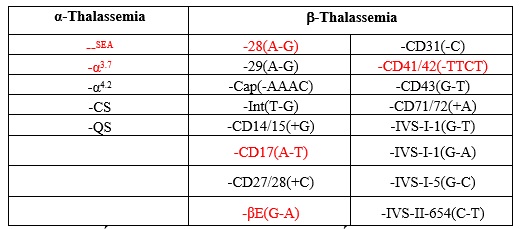
Bảng 2: 5 loại đột biến Alpha Thalassemia và 16 loại đột biến Beta Thalassemia thực hiện tại GENTIS. Các đột biến tô đỏ thường xuất hiện ở quần thể người Việt (theo số liệu GENTIS 2017-2019).
Tổng hợp: Nguyễn Thị Minh Lý –
Trung tâm xét nghiệm GENTIS HCM
Các chủ đề được trình bày sau chủ đề này:
- Hemogolobin và các bệnh liên quan
- Bệnh Thalassemia (các dạng và cấu trúc của anpha Thalassemia và Beta Thalassemia)
- Vai trò của việc tầm soát sớm Thalassemia, các phương pháp chẩn đoán
- Phụ nữ Thalassemia mang thai: Thách thức và giải pháp
Nguồn tham khảo:
- Antonio Cao, Renzo Galanello, Beta-Thalassemia, Genetics in Medicine Vol 12, Tr. 61–76 (2010).
- Dharmesh Chandra Sharma, Anita Arya, Purnima Kishor, Poonam Woike, Jyoti Bindal, Overview on Thalassemias: a Review article, Medico Research Chronicles, 2017, ISSN No. 2394-3971.
- P. Lahiri, S.A. Al-Attar, R.A. Hegele, Understanding Beta-Thalassemia with Focus on the Indian Subcontinent and the Middle East, The Open Hematology Journal, 2008, 2, 5-13.
- https://danso.org/viet-nam/
- https://www.middleeastmedicalportal.com/disorder-of-Thalassemias-and-hemoglobinopathies-a-genetic-overview/



Để lại bình luận
0 Bình luận