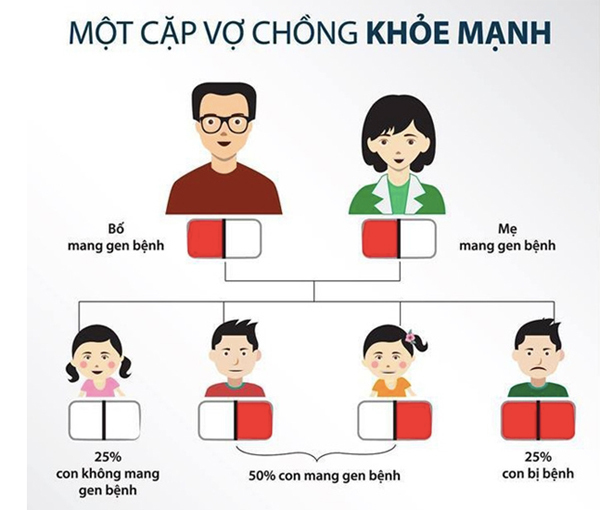
Có 7% người dân trên toàn cầu mang gen bệnh tan máu bẩm sinh; 1,1% cặp vợ chồng có nguy cơ sinh con bị bệnh hoặc mang gen bệnh.
Theo báo cáo tại hội nghị Thalassemia Châu Á Thái Bình Dương lần thứ 2 (năm 2015) Việt Nam hiện là một trong những quốc gia có tỉ lệ người mang gen bệnh Thalassemia cao nhất khu vực với khoảng 13% dân số, tương đương 10 triệu người, trong đó có khoảng 20.000 người ở thể nặng. Hậu quả, mỗi năm Việt Nam có thêm 2.000 trẻ sinh ra mắc Thalassemia trên tổng số 15.000 trẻ mới sinh mắc căn bệnh này trên toàn thế giới. Bệnh có biểu hiện đa dạng tùy kiểu đột biến gen, đối với các trường hợp thể nặng có thể gây tử vong ngay từ bào thai hoặc phải truyền máu, thải sắt suốt đời gây gánh nặng về kinh tế cho gia đình và xã hội.
Tuy nhiên bệnh Thalassemia hoàn toàn có thể dự phòng được thông qua chẩn đoán tiền hôn nhân và tiền sinh sản. Do đó, vấn đề tầm soát bệnh Thalassemia tiền hôn nhân, sàng lọc nguy cơ gen Thalassemia ở người nam và nữ trước khi kết hôn trở nên rất quan trọng để góp phần giảm thiểu tỷ lệ trẻ mang bệnh Thalassemia sinh ra mỗi năm.

Phác đồ chẩn đoán bệnh Thalassemia
1. Xét nghiệm tổng phân tích tế bào máu ngoại vi:
Các chỉ số cần đánh giá:
– Nồng độ hemoglobin (Hb)
– Lượng hemoglobin trung bình trong một hồng cầu (MCH)
– Thể tích trung bình hồng cầu (MCV).
- Thalassemia thể nhẹ: Có thể nghi ngờ thalassemia thể nhẹ (người lành mang gen bệnh) nếu có thiếu máu nhược sắc, hồng cầu nhỏ (3 chỉ số nêu trên đều giảm nhẹ). Bệnh lý này thường bị nhầm với thiếu máu thiếu sắt do MCV và MCH đều giảm.
- Thalassemia thể nặng: Nồng độ hemoglobin dao động trong khoảng 2-8 g/dl. Thể tích trung bình hồng cầu (MCV) và hemoglobin trung bình trong một hồng cầu (MCH) rất thấp. Số lượng bạch cầu thường tăng, do quá trình tan máu. Tiểu cầu có thể giảm nếu lách to đáng kể.
Đối với bệnh nhân thalassemia, hồng cầu thường nhợt nhạt, kích thước nhỏ. Hình dáng hồng cầu có thể bị biến đổi nặng nề, hồng cầu to nhỏ không đều, xuất hiện các hình dạng bất thường: hình nhẫn, hình bia, hình giọt nước. Hồng cầu bắt màu không đều: chỗ sẫm, chỗ nhạt.
2. Làm các xét nghiệm chuyên sâu để chẩn đoán xác định bệnh Thalassemia: Điện di huyết sắc tố và xét nghiệm gen Thalassemia
Ý nghĩa của Điện di huyết sắc tố và xét nghiệm Gen trong chẩn đoán bệnh THALASSEMIA
Cả 2 xét nghiệm đều giúp chẩn đoán bệnh Thalassemia, tuy nhiên trong khi xét nghiêm điện di đưa ra kết quả phát hiện sự xuất hiện của các thành phần Hemoglobin bất thường (sự biểu hiện bệnh về mặt huyết học) thì xét nghiệm gen sẽ xác định chính xác đột biến gen tổng hợp chuỗi globin (nguyên nhân di truyền) gây bệnh Thalassemia. Kết quả của xét nghiệm gen sẽ đưa ra là đột biến dạng đồng hợp tử hay dị hợp tử , từ đó ngoài việc chẩn đoán xác định thể bệnh Thalassemia còn có ý nghĩa trong việc tư vấn di truyền bệnh cho thế hệ sau.
 XÉT NGHIỆM ĐIỆN DI HUYẾT SẮC TỐ
XÉT NGHIỆM ĐIỆN DI HUYẾT SẮC TỐ
 XÉT NGHIỆM ĐIỆN DI HUYẾT SẮC TỐ
XÉT NGHIỆM ĐIỆN DI HUYẾT SẮC TỐĐiện di huyết sắc tố được dùng như một xét nghiệm đầu tay để tiếp cận một trường hợp nghi ngờ bệnh Thalassemia.
Kỹ thuật điện di dựa trên nguyên tắc là các phân tử tích điện hòa tan hay phân tán trong chất điện giải sẽ di chuyển khi có dòng điện đi qua. Cation di chuyển về phía cực âm, anion di chuyển về phía cực dương, các phân tử không mang điện tích sẽ không di chuyển. Như vậy, sau khi điện di, các thành phần Hb khác nhau trong mẫu máu sẽ được tách ra, từ đó có thể định danh và định tỉ lệ % của loại Hb đó trong mẫu thử. Có nhiều phương pháp điện di Hb như: điện di trong môi trường pH kiềm trên giấy acetat cellulose, điện di trên gel agar trong môi trường pH acid, điện di đẳng điện trên gel agarose hay gel polyacrylamide, điện di mao quản …
Đối với bệnh nhân Thalassemia: có thể phát hiện các hemoglobin bất thường: hemoglobin bào thai (HbF) trong hồng cầu của bệnh nhân β thalassemia, HbH trong bệnh HbH (α thalassemia thể vừa) hoặc Hb Bart’s trong α thalassemia thể nặng.
Thay đổi thành phần Hb trên điện di là đặc hiệu cho chẩn đoán β Thalassemia nặng:
- Hb F tăng cao > 10-90%.
- Hb A2 tăng >3,5%.
- Hb A1 giảm nặng hoặc không có.
XÉT NGHIỆM GEN GÂY BỆNH THALASSEMIA
Thalassemia là một nhóm bệnh thiếu máu tan máu bẩm sinh do đột biến gen globin gây nên thiếu hụt tổng hợp một hay nhiều mạch polypeptid trong globin của hemoglobin.
Các gen mã hóa cho sự tổng hợp các thành phần globin của hemoglobin người được sắp xếp thành 2 cụm (cluster). Các gen loại α thấy ở trên nhiễm sắc thể 16, còn gen-loại β ở trên nhiễm sắc thể 11. Tùy theo sự thiếu hụt tổng hợp ở mạch alpha, beta mà gọi là thể bệnh α-thalassemia hoặc β-thalassemia.
α-thalassemia
Bệnh α-thalassemia xuất hiện ở tất cả các chủng tộc trên thế giới, rất phổ biến ở các nước Đông Nam Á. Hiện có khoảng 5% dân số thế giới là người mang gen bệnh α-thalassemia, bao gồm dạng α+-thalassemia, α0-thalassemia, phân bố khác nhau ở từng khu vực, quốc gia, chủng tộc khác nhau [1]. Tại Trung Quốc, người mang gen α-thalassemia chiếm 5-15% dân số [2], Hong Kong 4% [3], Thailand 15-30% [4], Lào 43% [5], Việt Nam 5% [6]. Người bình thường có hai gen α globin nằm trên mỗi NST 16, và có tổng số bốn gen α globin trên hai NST 16 tương đồng (αα/αα). Tùy theo số lượng gen α bị đột biến, và tùy theo sự kết hợp đa dạng giữa các dạng allen đột biến khác nhau của bệnh α-thalassemia, gây ra các biểu hiện lâm sàng ở nhiều mức độ khác nhau. Bệnh Hemoglobin H (HbH) là thể trung gian của α-thalassemia, trong đó ba trên bốn gen α globin bị đột biến [7].
Trẻ mắc bệnh HbH thường có thiếu máu tan máu, có thể phải phụ thuộc truyền máu, gây hậu quả nghiêm trọng cho hàng loạt các cơ quan trong cơ thể. Nếu không được điều trị, trẻ mắc HbH thể nặng thường tử vong sớm, hoặc muộn hơn vì các biến chứng của bệnh.
Hội chứng phù thai do Hb Bart’s là thể nặng nhất của bệnh α-thalassemia, do đột biến mất hoàn toàn bốn gen α globin, gây thiếu máu nặng, dẫn đến suy tim, tràn dịch đa màng, phù toàn thân, thường chết lưu trong khoảng từ 28-40 tuần, hoặc tử vong ngay trong vài giờ đầu sau khi sinh. Ngoài ra, hội chứng phù thai do Hb Bart’s còn làm tăng nguy cơ nhiễm độc thai nghén và tiền sản giật cũng như các biến chứng sản khoa khác cho sản phụ
90% bệnh αthalassemia do đột biến mất đoạn trên một gen hoặc cả hai gen α globin, hoặc toàn bộ cả cụm gen α globin. Ngoài ra, có 10% không do đột biến mất đoạn gen, mà thường là các đột biến điểm trên gen α globin gây nên. Các đột biến mất đoạn hoặc không mất đoạn này sẽ tạo ra các allen đột biến dạng α0 và α+ -thalassemia [1].
Các loại allen đột biến của bệnh α-thalassemia: -αSEA; -α3.7; - α4.2; -CS; -QS, -Αmed, -Αthal, -Αfil.
β-thalassemia
Các nghiên cứu được thực hiện tại Việt Nam đều cho thấy có 8 loại đột biến gây ra 95% các trường hợp β-thalassemia ở người Việt Nam, gồm Cd17 (AAG-TAG), CD41/42 (-TCTT), -28 (A>G), CD71/72 (+A), IVSI-1 (G>T), IVSI-5 (G>C), IVSII-654 (C>T) và CD26 (GAG>AAG) gây bệnh HbE [8] [9] [10].
Biểu hiện lâm sàng và huyết học của bệnh β-thalassemia rất không đồng nhất, khác nhau tùy thể lâm sàng, tùy theo mức độ nặng, nhẹ của bệnh. Sở dĩ như vậy vì bệnh sinh cơ bản của bệnh β-thalassemia là mức độ mất cân bằng tổng hợp mạch globin β, do sự tương tác của các đột biến gen β-globin. Bệnh nhân β-thalassemia có biểu hiện lâm sàng rất đa dạng từ thiếu máu nhẹ cho tới các trường hợp nặng là thiếu máu Cooley gây nhiều biến chứng nguy hiểm và phụ thuộc truyền máu.
Có thể thấy việc kết hợp cả xét nghiệm huyết sắc tố cùng với xét nghiệm gen gây bệnh Thalassemia sẽ giúp Bác sĩ đưa ra được kết luận chính xác nhất về thể bệnh, mức độ biểu hiện lâm sàng cũng như tư vấn di truyền cho bệnh nhân.
Tài liệu tham khảo:
- John O., Joanne T.S., Renzo G., et al. (2013). Prevention of Thalassemia and other Hemoglobin Disorders. Thalassemia international federation publications
- He S., Zhang Q., Li D., et al. (2014). Prevention and control of Hb Bart's disease in Guangxi Zhuang Autonomous Region, China. Eur J Obstet Gynecol Reprod Biol, 178(7)-138-41
- Chan V., Chan T.K., Tang M., et al. (1995). Prenatal diagnosis and screening of common genetic diseases in Hong Kong. Southeast Asian J Trop Med Public Health, 26(1)-1-2
- Boonsa S., Sanchaisuriya K., Fucharoen G., et al. (2004). The diverse molecular basis and hematological features of Hb H and AEBart's diseases in Northeast Thailand. Acta Haematol, 111(3)-149-54
- Fucharoen S. and Winichagoon P. (2011). Haemoglobinopathies in southeast Asia. Indian J Med Res, 134(6)-498-506
- Trí Nguyễn Anh (2012). Viet Nam - Current Situation in Control Strategies and Health Systems in Asia. Health and Medicine,
- Fucharoen S. and Winichagoon P. (1992). Thalassemia in SouthEast Asia: problems and strategy for prevention and control. Southeast Asian J Trop Med Public Health, 23(4)-647-55
- Lê Thị Hảo, Pissard S, Van PH và cs (2001). Molecular analysis of βthalassemia in South Vietnam Hemoglobin 25: 305-309.
- Saovaros S, Trần Minh Hiếu, Thongperm M và cs(2002). Molecular analysis of β-thalassemia in South Vietnam, J of Hemoglobin 71: 85-88.
- Trần Văn Bé, Trần Minh Hiếu (2003). Phát hiện 8 đột biến gây bệnh βthalassemia ở Đông Nam Á bằng phương pháp ASO. Y học Việt Nam 2003: 1-5