-1366x0.jpg)
Hậu quả của bệnh Thalassemia:
Tùy theo dạng đột biến sẽ gây ra những biến chứng từ nhẹ (không ảnh hưởng nhiều đến sức khỏe nhưng có thể di truyền cho thế hệ sau) đến những biến chứng trung gian và nặng.
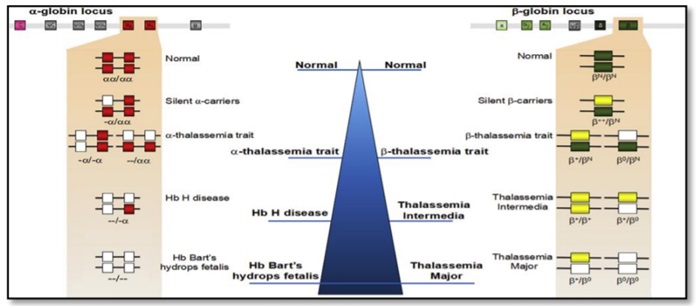
Hình 1: Phân loại các mức độ lâm sàng và biểu hiện kiểu gen - kiểu hình của các thể Alpha Thalassemia và Beta Thalassemia. Trong đó các locus gen Alpha nằm trên nhiễm sắc thể (NST) 16, các locus gen Beta nằm trên NST 11. Hình tam giác thể hiện các mức độ khác nhau (đi từ trên đỉnh xuống đáy thể hiện mức độ từ bình thường đến nặng). Các dạng alpha Thalassemia gồm: bình thường (normal); thể ẩn (Silent - carriers); thể nhẹ (-trait); thể Hb H; thể nặng (Hb Bart’s hydrops fetalis - phù thai). Các dạng Beta Thalassemia gồm: bình thường (normal); thể ẩn (Silent carriers); thể nhẹ (-trait); thể trung gian (Intermedia); thể nặng (major).
Những người bị bệnh Thalassemia có nguy cơ nhiễm trùng cao hơn. Điều này đặc biệt đúng nếu lá lách đã được cắt bỏ (do sự mở rộng của lá lách). Bệnh Thalassemia có thể làm cho tủy xương mở rộng, khiến xương mở rộng. Điều này có thể dẫn đến cấu trúc xương bất thường, đặc biệt là ở mặt và hộp sọ. Sự mở rộng tủy xương cũng làm cho xương mỏng và giòn, làm tăng nguy cơ gãy xương. Lách to có thể làm cho tình trạng thiếu máu trở nên tồi tệ hơn, và nó có thể làm giảm tuổi thọ của các tế bào hồng cầu được truyền máu. Thiếu máu có thể khiến trẻ tăng trưởng chậm. Tuổi dậy thì cũng có thể bị trì hoãn ở trẻ em mắc bệnh Thalassemia. Bệnh tim, chẳng hạn như suy tim sung huyết và nhịp tim bất thường, có thể liên quan đến bệnh Thalassemia nặng.

Nếu cả cha và mẹ đều có mang gen bệnh (thể ẩn) thì xác suất con bị bệnh Thalassemia lên đến 25%.
Quá tải sắt là biến chứng có liên quan nhất đến điều trị truyền máu. Khi bệnh nhân dùng chế độ truyền máu thường xuyên sẽ phát triển dần dần các biểu hiện lâm sàng của tình trạng quá tải sắt: suy sinh dục (35-55% bệnh nhân), suy giáp (9-11%), suy tuyến cận giáp (4%), tiểu đường (6- 10%), gan xơ hóa và rối loạn chức năng tim (33%).

Các hậu quả của beta Thalassemia
Chẩn đoán Thalassemia
Một số xét nghiệm có thể được sử dụng để giúp phát hiện và chẩn đoán bệnh Thalassemia:
| Loại xét nghiệm: Complete blood count (CBC) Công thức máu Mục đích: - Đánh giá các tế bào trong máu - Xác định chỉ số hồng cầu Dựa trên các chỉ tiêu: - Thể tích trung bình (MCV) -Số lượng hồng cầu và lượng Hb có trong đó - Kích thước, hình dạng |
| Loại xét nghiệm: Blood smear (phết tế bào ngoại biên) Mục đích: Đánh giá dưới kính hiển vi xem số lượng và các loại tế bào bạch cầu, hồng cầu và tiểu cầu xem chúng có bình thường và trưởng thành hay không Dựa trên các chỉ tiêu: - Trở nên nhạt hơn bình thường (hypochromic) - Thay đổi kích thước và hình dạng (anisocytosis và poikilocytosis) - Có sự phân bố huyết sắc tố không đồng đều |
| Loại xét nghiệm: Iron studies (gồm các xét nghiệm đo lường việc lưu trữ và sử dụng sắt của cơ thể) Mục đích: Đánh giá sắt, ferritin, khả năng liên kết sắt chưa bão hòa (UIBC), tổng công suất liên kết sắt (TIBC). Dựa trên các chỉ tiêu: - UIBC - TIBC |
| Loại xét nghiệm: Điện di Hemoglobin Mục đích: Đánh giá loại và định lượng tương đối của huyết sắc tố có trong hồng cầu. Dựa trên các chỉ tiêu: So sánh chỉ số Hb tiêu chuẩn ở người trưởng thành: - Hb A: 95-98% - Hb A2: 2-3% - Hb F: <2% |
| Loại xét nghiệm: DNA analysis (Phân tích DNA) Mục đích: - Xác nhận các đột biến trong các gen sả n xuất alpha và beta globin. - Family studies (nghiên cứu sự xuất hiện bệnh của các thành viên trong gia đình để hạn chế di truyền cho các thế hệ sau) - Xét nghiệm nước ối: được sử dụng cho những trường hợp thai nhi có nguy cơ mắc Thalassemia. Dựa trên các chỉ tiêu: - Anpha Thalassemia: phát hiện đột biến ở 2 gen HBA1, HBA2. - Beta Thalassemia: phát hiện đột biến ở gen HBB. |
Nhìn chung, các bước chẩn đoán của bệnh Thalassemia bao gồm sự xác nhận ban đầu của bệnh là rối loạn Thalassemia và sự khác biệt của nó với các rối loạn tổng hợp Hemoglobin bẩm sinh và mắc phải khác có thể bắt chước các hội chứng Thalassemia.
- Ở trạng thái silent carrier (thể ẩn) bệnh nhân mắc bệnh Thalassemia về cơ bản không có triệu chứng và CBC, điện di Hemoglobin và phết tế bào ngoại biên thường là bình thường.
- Dạng Hypochromia và Microcytosis (tế bào hồng cầu nhạt hơn) nhẹ có thể được đánh giá bằng kính hiển vi.
- Ở bệnh thiếu máu alpha Thalassemia, tế bào hồng cầu là bất thường với các dạng Microcytosis, Hypochromia và tăng lượng Hb Bart được ghi nhận (3% -8%). Hội chứng Hydrops Fetalis (phù thai) được công nhận bằng cách phát hiện một trẻ sơ sinh phù, thiếu máu nặng và sự hiện diện của 80% Hb Barts trên điện di Hemoglobin.
- Các đồng hợp tử dạng beta Thalassemia nặng dễ dàng nhận ra bởi sự thay đổi huyết học với mức Hb F, Hb A2 rất cao. Các trạng thái dị hợp tử được xác nhận bởi các tế bào hồng cầu hypocromic vi mô và mức độ Hb A2 tăng cao.
Các xét nghiệm phân tích DNA được sử dụng để giúp xác nhận các đột biến trong các gen sản xuất alpha và beta globin. Xét nghiệm DNA không được thực hiện thường xuyên nhưng có thể được sử dụng để giúp chẩn đoán bệnh Thalassemia và để xác định tình trạng mang mầm bệnh, nếu được chỉ định.
Xét nghiệm phân tử cho alpha Thalassemia phát hiện các đột biến phổ biến (ví dụ: mất đoạn) trong hai gen alpha HBA1 và HBA2. Mỗi người có hai bản sao của mỗi gen này (được gọi là alen), trong các tế bào của chúng thì một nhận từ mẹ và một nhận từ cha. Những alen này chi phối việc sản xuất alpha globin và nếu đột biến dẫn đến mất chức năng của một hoặc nhiều gen alpha.
Đối với beta Thalassemia, gen beta Hemoglobin -HBB, có thể được phân tích hoặc giải trình tự để xác nhận sự hiện diện của các đột biến gây ra bệnh Thalassemia. Các xét nghiệm di truyền cũng có thể được đưa ra cho các đột biến HBB khác như đột biến Hb S, có liên quan đến bệnh hồng cầu hình liềm. Hơn 250 đột biến có liên quan đến beta Thalassemia, mặc dù một số nguyên nhân không có dấu hiệu hoặc triệu chứng. Sự hiện diện của một trong những đột biến đó xác nhận chẩn đoán beta Thalassemia.
Xét nghiệm Thalassemia có thể thực hiện với vài ml máu
Xét nghiệm di truyền nước ối được chỉ định trong những trường hợp hiếm hoi, thai nhi có nguy cơ mắc bệnh Thalassemia. Điều này đặc biệt quan trọng nếu cả hai cha mẹ có khả năng mang đột biến vì điều đó làm tăng nguy cơ con họ có thể thừa hưởng sự kết hợp của các gen bất thường, gây ra một dạng bệnh Thalassemia nặng hơn.
Điều trị Thalassemia
Hầu hết các cá nhân có đặc điểm Thalassemia nhẹ không cần điều trị. Tuy nhiên, họ có thể muốn xem xét tư vấn di truyền, vì họ có thể truyền gen đột biến cho con cái của họ.
Hb Bart hydrops, Hội chứng phù thai hoặc alpha Thalassemia Major hiện không có cách điều trị hiệu quả và trẻ thường bị sảy thai, chết non hoặc chết ngay sau khi sinh. Nỗ lực truyền máu trong tử cung, sau khi phát hiện sớm trước sinh bằng siêu âm Doppler của tình trạng này, đã được tiến hành, nhưng hầu hết những thai nhi sống sót đều trải qua tỷ lệ dị tật bẩm sinh cao và các nỗ lực nên được khuyến khích cho đến khi điều trị hiệu quả hơn (ví dụ như sử dụng liệu pháp gen).
Những người mắc bệnh Hemoglobin H hoặc beta Thalassemia trung gian sẽ trải qua số lượng thiếu máu khác nhau trong suốt cuộc đời của họ. Họ có thể sống cuộc sống tương đối bình thường nhưng sẽ cần theo dõi thường xuyên và đôi khi có thể cần truyền máu. Bổ sung axit folic thường được đưa ra, nhưng bổ sung sắt không được khuyến khích.
Điều trị cho những người bị bệnh Thalassemia trung gian là hạn chế các triệu chứng. Vì cường lách có thể gây ra tình trạng thiếu máu ngày càng trầm trọng, chậm phát triển và rối loạn cơ học từ lá lách lớn, cắt lách là một khía cạnh liên quan của việc kiểm soát bệnh Thalassemia trung gian.

Do tỉ lệ cao người mang gen bệnh (thể ẩn) trong quần thể, mọi cá nhân chuẩn bị kết hôn đều được khuyến khích thực hiện tầm soát bệnh Thalassemia
Cho đến ngày nay, Ghép tủy xương (BMT) vẫn là phương pháp điều trị dứt điểm có sẵn cho bệnh nhân mắc bệnh Thalassemia. BMT thành công đầu tiên được thực hiện vào những năm 1980 bởi Giáo sư Guido Lucarelli. Ở những bệnh nhân trẻ tuổi nguy cơ thấp, tỷ lệ sống sót của Thalassemia là 87%, tỷ lệ tử vong là 3%. Hạn chế là phương pháp chữa bệnh này đòi hỏi người cho (donor) tương thích với kháng nguyên bạch cầu của người (HLA) với người nhận.
Nếu bệnh nhân không có người hiến phù hợp với HLA thì có một phương pháp chữa bệnh khác gọi là Ghép xương (BMT) từ người mẹ sang con (người hiến không khớp), trong đó người hiến là mẹ. Phương pháp được phát minh vào năm 2002 bởi bác sĩ Pietro Sodani. Kết quả là tỷ lệ sống 70%, thải ghép 23% và tỷ lệ tử vong 7%. Kết quả tốt nhất là với những bệnh nhân rất trẻ [61].
Liệu pháp gen cho beta Thalassemia vẫn đang được thử nghiệm và hy vọng cho tương lai. Việc theo dõi và điều trị hiện tại dành cho phần lớn bệnh nhân beta Thalassemia thể nặng (major) ở các nước có nền kinh tế xã hội thấp là truyền máu thường xuyên; điều trị thải sắt và quản lý các biến chứng của quá tải sắ. Mục đích của liệu pháp truyền máu là điều chỉnh bệnh thiếu máu và duy trì mức độ lưu thông của huyết sắc tố (Hb) đủ để ức chế hồng cầu nội sinh.
Có thể tóm tắt các phương pháp điều trị theo bảng sau:
| Truyền thống (thông thường) | Không truyền thống |
| Truyền máu (Blood Transfusions- BT) | Ghép tủy xương (BMT) |
| Liệu pháp thải sắt (Iron chelation Therapy- ICT) | Tế bào gốc tạo máu (hematopoietic stem) |
| Cắt lách (Splenectomy) | Liệu pháp tương lai (Future Therapy) |
| Liệu pháp hỗ trợ (Supportive therapy) | Cấy máu cuống rốn (Cord Blood Transplantation- CBT) |
| Hỗ trợ tâm lý (Psychological support) |
|
Tầm quan trọng của việc sàng lọc sớm Thalassemia:
Bệnh Thalassemia gây nhiều tổn thất cho gia đình và xã hội. Thalassemia là nỗi lo toàn cầu, thế giới bắt đầu lấy ngày 8/5/1986 đánh dấu cho các hoạt động đẩy lùi bệnh Thalassemia. Theo thống kê tại Việt Nam, chi phí cho xét nghiệm 1 ca Thalassemia từ khi sinh ra đến 30 tuổi trung bình tầm 3 tỷ đồng. Tuy nhiên, việc kiểm soát và tầm soát bệnh (như tầm soát tiền hồn nhân để giảm nguy cơ trẻ mắc bệnh ra đời) lại rất thấp. Phù hợp với mức thu nhập của đại đa số dân số Việt Nam). Ví dụ chi phí cho việc xét nghiệm ADN Thalassemia dao động khoảng tầm hơn 2 triệu/trường hợp. Chi phí cho việc hỗ trợ sinh sản khi cả bố và mẹ mang gen bệnh muốn sinh con khỏe mạnh bằng phương pháp sàng lọc PGD (preimplantation genetic screening- sàng lọc di truyền trước chuyển phôi) giờ đây cũng rất hợp lý.
GENTIS thực hiện Sàng lọc phôi trước khi chuyển khi làm IVF đã giúp nhiều cặp vợ chồng mang gen Thalassemia có con khỏe mạnh
Tại GENTIS, nhiều cặp vợ chồng mắc Thalassemia (phổ biến là các thể dạng alpha SEA, anpha 3.7; beta E (G-A); beta 28 (A-G)) đã tiến hành các xét nghiệm PGD. Kết quả đã chọn được các phôi (sau thụ tinh IVF) có tỷ lệ sống cao, không mắc bệnh Thalassemia hoặc ở dạng nhẹ hơn so với bố mẹ. Điều đặc biệt hơn nữa, với xét nghiệm PGD đã mang nhiều cơ hội và hy vọng cho các cặp vợ chồng sinh con đầu trước đó mắc bệnh nhưng các bé sau hoàn toàn bình thường.
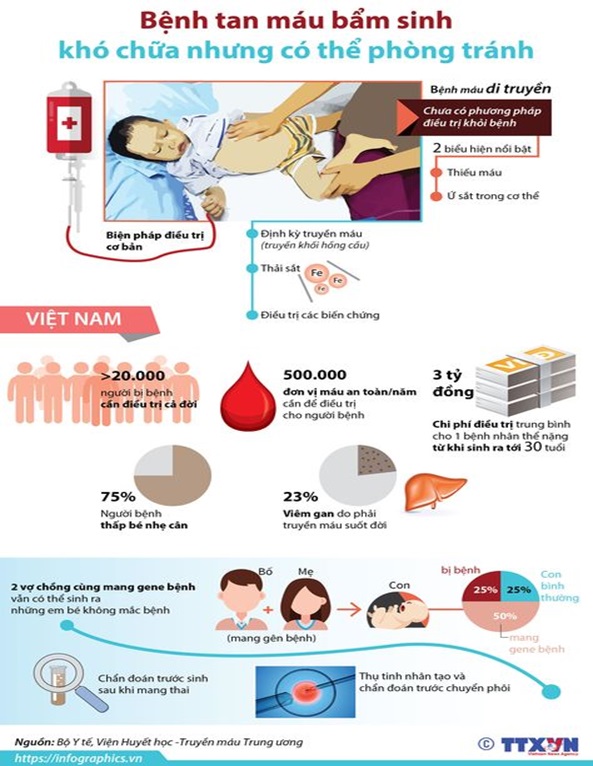
Hình 3: Hậu quả của Thalassmia và các phương pháp ngăn ngừa di truyền cho các thế hệ sau
Tổng hợp: Nguyễn Thị Minh Lý
Trung tâm xét nghiệm GENTIS HCM
Các chủ đề được trình bày sau chủ đề này:
- Hemogolobin và các bệnh liên quan
- Bệnh Thalassemia (các dạng và cấu trúc của anpha Thalassemia và Beta Thalassemia)
- Vai trò của việc tầm soát sớm Thalassemia, các phương pháp chẩn đoán
- Phụ nữ Thalassemia mang thai: Thách thức và giải pháp
Nguồn tham khảo:
- Aisha Ibrahim Al Naami, Dr. Ohood Othman Felemban, Disease Knowledge and Treatment Adherence Among Thalassemia Patients: A Literature Review, IOSR Journal of Nursing and Health Science, e-ISSN: 2320–1959.p- ISSN: 2320–1940 Volume 8, Issue 3 Ser. III. (May. - June .2019), PP 70-77.
- Dharmesh Chandra Sharma, Anita Arya, Purnima Kishor, Poonam Woike, Jyoti Bindal, Overview on Thalassemias: a Review article, Medico Research Chronicles, 2017, ISSN No. 2394-3971.



Để lại bình luận
0 Bình luận